


Advantages
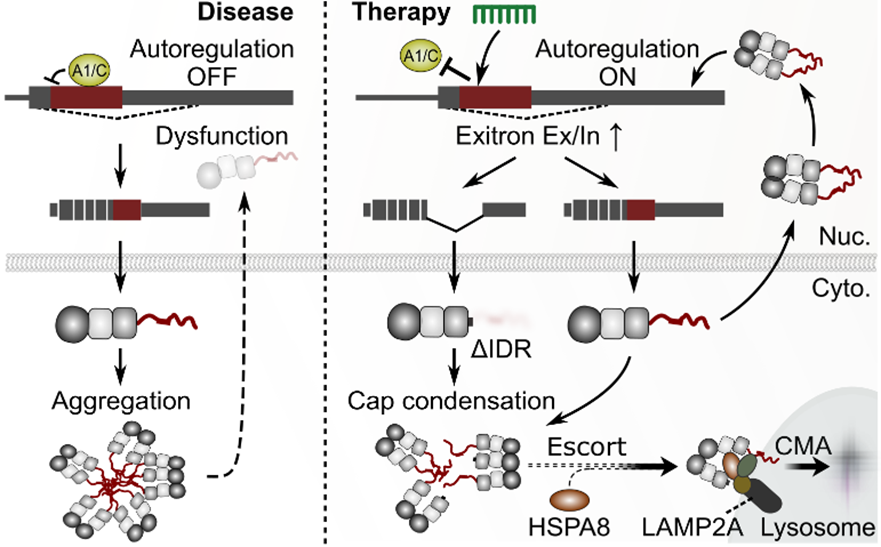
- A “Self-Repair” Concept that Corrects the TDP-43 Pathological Loop Upstream: Unlike simple expression suppression or downstream compensation, this strategy implements a two-layered defense. It controls exitron splicing to reduce the IDR load and utilizes the induced ΔIDR to contain aggregation. This breaks the vicious cycle of cytoplasmic aggregation and nuclear dysfunction, restoring TDP-43 homeostasis.
- Broad Applicability, Including Sporadic Cases: The underlying TDP-43 pathology is common to approximately 97% of ALS cases. This represents a cross-pathology targeting strategy that can also be expanded to TDP-43 type FTLD.
 |
Current Stage & Key Data
In vivo Proof of Concept (POC) in disease model animals is complete, and the project is in the preclinical preparation stage for GLP studies. The university is leading the foundational work, with the expectation of transitioning to company-led preclinical trials.
- Suppression of Aggregation and Functional Recovery (In Vitro): In a human neuronal cell model (siCSE1L) mimicking ALS-like pathology (nuclear leakage and dysfunction), an ASO (CS3) targeting the HNRNPC binding site was introduced. This suppressed the abnormal accumulation of cytoplasmic TDP-43 and significantly restored the expression/splicing of nuclear function markers, including STMN2.
- Therapeutic Effects in an ALS Mouse Model (In Vivo): In an ALS mouse model exhibiting TDP-43 aggregation and motor neuron degeneration (Rpt3 CKO mouse), an ASO (AS5) targeting the HNRNPA1 binding site was administered intrathecally. In addition to suppressing abnormal TDP-43 aggregation and improving neuronal survival, the treatment suppressed the decline in motor function (grip strength) and significantly extended the lifespan (log-rank p=0.011).
Partnering Model
Co-development or licensing agreements are envisioned, based on the university’s POC data and patent portfolio. The partner company would lead all processes from the GLP studies onward, while the university provides scientific support regarding the mechanism of action, pathology evaluation, and biomarkers. This phased technology transfer is designed to optimize risks and timelines.。
Technology Overview & Background
Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Lobar Degeneration (FTLD) are proteinopathies in which nuclear dysfunction and cytoplasmic aggregation of TDP-43 occur concurrently. TDP-43 self-regulates its output by splicing out an “exitron” (an intron within TARDBP exon 6) to adjust the amount of sequence containing the IDR. In the state of the disease, this “switch” is turned OFF, accelerating a vicious cycle of IDR excess, which promotes aggregation and leads to nuclear depletion. This ASO sterically blocks HNRNP A1/C binding to turn the exitron splicing “switch” back ON, restoring TDP-43 homeostasis through a two-layered mechanism:
Layer 1: Reduction of IDR Load (Suppresses the production of aggregation-prone full-length TDP-43)
Layer 2: Induction of the ΔIDR Isoform (Caps aggregation and escorts it for degradation via Chaperone-Mediated Autophagy (CMA) mediated by HSPA8–LAMP2A)
A key strength of this technology is its ability to intervene in the core of pathology, regardless of genetic background.
Patents & Publications
WO2022/113799, WO2023/204313, and multiple other patent applications are pending (undisclosed).
The description of the mechanism of action and efficacy data is based on information publicly available in preprints and conference presentations.
Principal Investigator & Academic Institution
Akihiro Sugai (Niigata University)
Project ID:WL-05310