


Advantage and Core Benefit
- Incorporating local distance correction into recurrence plots enables more accurate reconstruction of 3D chromosomal structures from single cell Hi-C data.
- No additional experimental steps are required, and the method can be seamlessly integrated into existing single cell Hi-C analysis workflows.
Background and Technology
The three-dimensional (3D) structure of chromosomes plays a crucial role in understanding the physical interactions between DNA and proteins and is deeply involved in epigenomic regulation and gene expression. Among the established approaches to analyzing chromatin structure, the Hi-C (genome-wide chromatin conformation capture) technique has been widely used. Hi-C works by crosslinking spatially proximal DNA regions in the nucleus, digesting and ligating them with restriction enzymes, and sequencing the resulting fragments to infer 3D chromatin structure. By single cell Hi-C data, we can learn such crosslinking in each cell. Typically, reconstruction involves generating a low-resolution model, which is then refined to obtain a high-resolution structure. However, the quality of the initial low-resolution model significantly affects the accuracy of the final output, highlighting the need for more precise reconstruction methods at early stages.
To address this challenge, Professor Yoshito Hirata of the University of Tsukuba has developed a novel approach that applies recurrence plots (RPs) a mathematical tool used in time-series analysis—to single cell Hi-C data. RPs are well-suited to visualizing patterns and distance relationships and are effective for structure reconstruction based on distance matrices. By incorporating corrections to local distances within the recurrence plot, the proposed method achieves superior reconstruction accuracy compared to conventional techniques. Importantly, this improvement is accomplished solely through computational processing of existing single cell Hi-C data, without any additional experimental steps. The method can thus be easily implemented into existing single cell Hi-C analysis pipelines. This advancement enhances the detection of critical structural domains such as Topologically Associating Domains (TADs) and supports further elucidation of biologically relevant genome organization.
Data
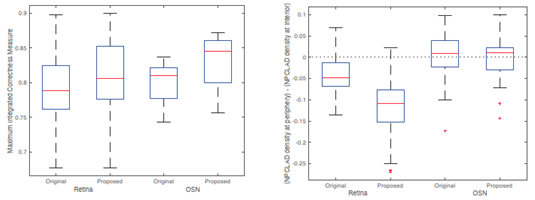
- The method was validated using publicly available single cell Hi-C datasets from retina and olfactory sensory neuron (OSN) cells. In both cases, the proposed method outperformed the original method in terms of the Integrated Correctness Measure, indicating higher reconstruction accuracy.
- Lamina Associated Domain (LAD) Distribution: Using neural progenitor cells (NPCs) as a reference, LAD positioning was evaluated in retina and OSN cells. In OSN cells, both original and proposed methods reproduced the outer-nuclear localization of LADs. In contrast, in retina cells, the proposed method better reproduced the known biological trend in which certain LADs shift toward the nuclear interior.
 |
Patent & Publication
Hirata et al., Scientific Reports, 2022 Jul 11; 12:11757. (DOI: 10.1038/s41598-022-15038-4)
WO/2023/048251
Researcher
Dr. Yoshito Hirata (University of Tsukuba)
Expectations
The University of Tsukuba is seeking partnerships with companies engaged in contract analysis or diagnostics services that are interested in applying this technology for precise 3D chromosomal structure inference. Technical evaluation and licensing discussions, including direct meetings with the researchers, are welcome. As part of a preliminary technical assessment, companies may also test the proposed method using their own single cell Hi-C datasets under confidentiality.
Project ID:WL-05096